WO2008106900A1 - Method of manufacturing (3r, 4s) -1- (4-fluorophenyl) -3- [ (3s) -3- (4 -fluorophenyl) -3-hydroxypropyl) ] -4- (4-hyd roxyphenyl) -2-azetidinone - Google Patents
Method of manufacturing (3r, 4s) -1- (4-fluorophenyl) -3- [ (3s) -3- (4 -fluorophenyl) -3-hydroxypropyl) ] -4- (4-hyd roxyphenyl) -2-azetidinone Download PDFInfo
- Publication number
- WO2008106900A1 WO2008106900A1 PCT/CZ2008/000023 CZ2008000023W WO2008106900A1 WO 2008106900 A1 WO2008106900 A1 WO 2008106900A1 CZ 2008000023 W CZ2008000023 W CZ 2008000023W WO 2008106900 A1 WO2008106900 A1 WO 2008106900A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- stage
- formula
- general formula
- temperature range
- water
- Prior art date
Links
- RGLDOOGSLUICQS-BTDUUNLVSA-N OC(C(CCC(c(cc1)ccc1F)=O)[C@H]1C(CC2)CC=C2O)N1c(cc1)ccc1F Chemical compound OC(C(CCC(c(cc1)ccc1F)=O)[C@H]1C(CC2)CC=C2O)N1c(cc1)ccc1F RGLDOOGSLUICQS-BTDUUNLVSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D205/00—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom
- C07D205/02—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom not condensed with other rings
- C07D205/06—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member
- C07D205/08—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member with one oxygen atom directly attached in position 2, e.g. beta-lactams
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Definitions
- the invention deals with a new method of manufacturing (3R,4S)-l-(4-fluorophenyl)- 3 - [(3 S)-3-(4-fluorophenyl)-3 -hydroxypropyl)] -4-(4-hydroxyphenyl)-2-azetidinone.
- ezetimibe is produced in such a way that ( ⁇ S)-4-hydroxybutanolide is added to iV-(4-benzyloxybenzylidene)-4- fluoroaniline with the use of LDA at -78 °C, the obtained diol is cleft with a periodate intG an aldehyde, which reacts with 4-fluoroacetophenone 0-trimethylsirylenol producing an aldol. The latter is dehydrated to produce an unsaturated ketone whose double bond or at the same time the protective benzyl group is hydrogenated on a palladium catalyst.
- the ketone is asymmetrically reduced with a diborane in the presence of a chiral ligand to produce ezetimibe or its O-benzyl derivative, which is hydrogenolyzed on a palladium catalyst.
- Disadvantages of this method consist in the necessity to work at very low temperatures and to use expensive catalysts of the palladium type.
- the production method of ezetimibe described in the US patent 5,856,473 is based on using 5-(4-fluorophenyl)-4-pentenoic acid, which is transformed to a chloride with the use of oxalylchloride and further to acyloxazolidide by reaction with ( ⁇ S)-4-phenyl-2-oxazolidmone.
- the latter is added onto N-(4-benzyloxybenzylidene)-4-fluoroaniline with the use of titanium tetrachloride in the presence of diisopropylethylamine to obtain a product that is cyclized with the use of bistrimethylsilylacetamide and catalytic TBAF to olefine-azetidinone.
- Glutaric acid methyl esterchloride is produced by the action of oxalylchloride on the corresponding acid and is reacted with ( ⁇ S)-4-phenyl-2-oxazolidinone to produce (S)-N-(4-methoxycarbonylbutanoyl)- oxazolidide.
- the latter is added onto the above mentioned N-(4-benzyloxybenzylidene)-4- fluoroaniline in the presence of titanium tetrachloride and the obtained product is cyclized by the action of bistrimethylsilylacetamide and catalytic TBAF to ester-azetidinone.
- This invention describes a new method of manufacturing (3R,4S)-l-(4-fluorophenyl)- 3 - [(3 S)-3 -(4-fluorophenyl)-3-hydroxypropyl)] -4-(4-hydroxyphenyl)-2-azetidinone (ezetir ⁇ ibe) of formula I
- R means a trialkylsilyl protective group, in the presence of a Lewis acid, e.g. titanium tetrachloride or isopropoxy titanium chloride, and a strong organic base in an inert organic solvent in the temperature range of -40 to 0 °C (stage 2), and the produced amino-oxazolidide of general formula VI
- R and n mean the same as above, is desilylated by the action of a desilylation agent such as tetrabutylammonium fluoride in an inert organic solvent in the temperature range of -10 to +40 0 C (stage 4), and thus obtained ketal of general formula VIII wherein n means the same as above, is deketalized by the action of acidic agents in a mixture of water and a water-miscible solvent in the range of temperatures of 0 to 80°C (stage 5), and the obtained ketone of formula IX
- ezetimibe can be produced with the use of a method that does not require the use of expensive catalysts of the palladium type, thus eliminating the risk of retention of small quantities of this metal in the produced substance.
- the production procedure consists of six stages, which are further described in a detailed way.
- the compound of general formula IV can also be prepared in such a way that oxazolidide of formula II is reacted with the bis(trimefhylsilyl) derivative of the alkyleneglycol in the presence of a catalyst, e.g. trimethylsilyl triflate.
- a catalyst e.g. trimethylsilyl triflate.
- the above mentioned bis(trimethylsilyl) derivative of alkyleneglycol can be obtained by sylilation of the alkyleneglycol of formula III with trimethylsilylchloride in the presence of a suitable base, e.g. triethylamine.
- a Lewis acid e.g. titanium tetrachloride or titanium isopropoxide chloride
- the addition is performed in the presence of a strong organic base, preferably diisopropylethylamine, in the quantity of 2 to 5 equivalents, in an inert organic solvent such as dichloromethane, dichloroethane, toluene, tert-butylmethylether, tetrahydrofuran, in the temperature range of -40 to 0 0 C, preferably at -35 to -20 0 C.
- a strong organic base preferably diisopropylethylamine
- an inert organic solvent such as dichloromethane, dichloroethane, toluene, tert-butylmethylether, tetrahydrofuran
- a silylation agent such as bis-(trimethylsilyl)acetamide
- a catalytic quantity of a fluoride preferably tetrabutylammonium fluoride.
- the cyclization is carried out in an inert organic solvent, such as tert-butylmethylether, tetrahydrofuran, toluene or dichloromethane, in the temperature range of -20 to 50 0 C, preferably at -5 to +10 0 C.
- the silylated azetidinone of general formula VII, wherein both R and n mean the same as above, is desilylated by the action of a desilylation agent, such as tetrabutylammonium fluoride, in an inert organic solvent, as e.g. tetrahydrofuran or tert- butylmethylether, in the temperature range of -10 to +40 0 C, preferably at -5 to +15 0 C.
- a desilylation agent such as tetrabutylammonium fluoride
- an inert organic solvent as e.g. tetrahydrofuran or tert- butylmethylether
- stage 5 In a beneficial alternative of the procedure the product of stage 3 is not isolated but desilylation is carried out immediately, as mentioned in stage 4 (one-pot transformation).
- Stage 5 The ketal of general formula VIII wherein n means the same as above is hydro lyzed by the action of acidic agents such as p-toluenesulfonic acid, methanesulfonic acid or acetic acid, in a mixture of water and a water-miscible solvent such as tetrahydrofuran, acetone, or isobutymethylketone, in the temperature range of 0 to 80 °C, beneficially at 50 to 70 °C.
- acidic agents such as p-toluenesulfonic acid, methanesulfonic acid or acetic acid
- a water-miscible solvent such as tetrahydrofuran, acetone, or isobutymethylketone
- the ketone of formula IX is reduced asymmetrically with the use of boron agents, such as e.g. a diborane, in the presence of the chiral ligand (i?)-2-methyl-CBS-oxazaborolidine in the quantity of 1 to 20 mol% in an inert organic solvent, e.g. tetrahydrofuran, tert- butylmethylether, toluene, or dichloromethane, in the temperature range of -20 to +40 °C.
- boron agents such as e.g. a diborane
- an inert organic solvent e.g. tetrahydrofuran, tert- butylmethylether, toluene, or dichloromethane
- the obtained compound of formula I (ezetimibe) is finally purified by crystallization from a mixture of water and an alcohol, e.g. 2-propanol or methanol.
- the mixture (dark wine-red solution) was stirred at that temperature for 2 h, and then 10% hydrochloric acid (50 ml) and toluene (70 ml) was added.
- a suspension of ketal of formula VIII (n 1) (2.63 g, 5.83 mmol) in a mixture of acetic acid (60 ml), THF (15 ml) and water (15 ml) was brought under stirring to 60 °C and kept at that temperature for 5 h, during which a homogeneous solution was formed. After completion of the reaction the mixture was concentrated on rotary evaporator and the concentrate was partitioned between toluene (100 ml) and saturated solution OfNaHCO 3 (30 ml). The separated organic layer was washed with saturated solution OfNaHCO 3 (Ix) and water (Ix) and, after drying (Na 2 SO 4 ), evaporated to dryness on rotary evaporator.
Abstract
A method of manufacturing (3R,4S)-l-(4-fluorophenyl)-3-[(3S)-3-(4-fluorophenyl)-3- hydroxypropyl)]-4-(4-hydroxyphenyl)-2-azetidinone (Ezetimibe) of formula I, starting from the optically active (S)-N-acyl-oxazolidide of formula II, which is reacted with an alkyleneglycol of general formula III, (stage 1), and the obtained acetal-oxazolidide of general formula IV, is subjected to reaction with a silyl-imine of general formula V the produced amino-oxazolidide of general formula VI, (stage 3), and the obtained silylated azetidinone of general formula VII, is desilylated (stage 4), and the ketal of general formula VIII produced this way, is deketalized formula IX is finally reduced.
Description
METHOD OF MANUFACTURING
(3R, 4S) -1- (4-FLUOROPHENYL) -3- [ (3S) -3- (4-FLUOROPHENYL) -3-HYDROXYPROPYL) ] -4- (4-HYD ROXYPHENYL) -2-AZETIDINONE
Technical Field
The invention deals with a new method of manufacturing (3R,4S)-l-(4-fluorophenyl)- 3 - [(3 S)-3-(4-fluorophenyl)-3 -hydroxypropyl)] -4-(4-hydroxyphenyl)-2-azetidinone.
Background Art
(3R,4S)- 1 -(4-Fluorophenyl)-3 - [(3 S)-3-(4-fluorophenyl)-3 -hydroxypropyl)] -4-(4- hydroxyphenyl)-2-azetidinone of formula I

lαiown under the INN name ezetimibe, is described in the US patent 5,631,365 as a hypo- lipidemic substance reducing intestinal absorption of cholesterol and other sterols.
In accordance with the US patents 5,739,321 and 5,886,171 ezetimibe is produced in such a way that (<S)-4-hydroxybutanolide is added to iV-(4-benzyloxybenzylidene)-4- fluoroaniline with the use of LDA at -78 °C, the obtained diol is cleft with a periodate intG an aldehyde, which reacts with 4-fluoroacetophenone 0-trimethylsirylenol producing an aldol. The latter is dehydrated to produce an unsaturated ketone whose double bond or at the same time the protective benzyl group is hydrogenated on a palladium catalyst. Then, the ketone is asymmetrically reduced with a diborane in the presence of a chiral ligand to produce ezetimibe or its O-benzyl derivative, which is hydrogenolyzed on a palladium catalyst. Disadvantages of this method consist in the necessity to work at very low temperatures and to use expensive catalysts of the palladium type.
The production method of ezetimibe described in the US patent 5,856,473 is based on using 5-(4-fluorophenyl)-4-pentenoic acid, which is transformed to a chloride with the use of oxalylchloride and further to acyloxazolidide by reaction with (<S)-4-phenyl-2-oxazolidmone.
The latter is added onto N-(4-benzyloxybenzylidene)-4-fluoroaniline with the use of titanium tetrachloride in the presence of diisopropylethylamine to obtain a product that is cyclized with the use of bistrimethylsilylacetamide and catalytic TBAF to olefine-azetidinone. This alkene is transformed to the ketone by the action OfPd(OAc)2 and benzoquinone in the presence of perchloric acid. The ketone is again asymmetrically reduced with a diborane in the presence of a chiral ligand and finally the protective (9-benzyl group is hydrogenolyzed. Repeated using of expensive catalysts of the palladium type and using the toxic oxalylchloride in the procedure are considerable disadvantages.
In accordance with the above mentioned US patent 5,631,365 ezetimibe is produced in such a way that (jS)-N-(4-methoxycarbonylbutanoyl)oxazolidide is synthesized from (S)-4- phenyl-2-oxazolidinone and glutaric acid esterchloride and then it is added onto the above mentioned N-(4-benzyloxybenzylidene)-4-fluoroaniline in the presence of titanium tetrachloride and the obtained product is cyclized by the action of bistrimethylsilylacetamide and catalytic TBAF to ester-azetidinone. Alkaline hydrolysis of the ester produces an acid, which is transformed to an acyl chloride whose reaction with a Grignard reagent in the presence OfZnCl2 and Pd(PPh3 )4 produces a ketone. The latter is asymmetrically reduced with a diborane in the presence of a chiral ligand and finally the protective O-benzyl group is hydrogenolyzed on a palladium catalyst. Again, repeated using of expensive catalysts of the palladium type as well as using the toxic oxalylchloride is a considerable disadvantages. The production method of ezetimibe in accordance with WO 2006/137080 is similar to the above mentioned one and it also has similar disadvantages. Glutaric acid methyl esterchloride is produced by the action of oxalylchloride on the corresponding acid and is reacted with (<S)-4-phenyl-2-oxazolidinone to produce (S)-N-(4-methoxycarbonylbutanoyl)- oxazolidide. The latter is added onto the above mentioned N-(4-benzyloxybenzylidene)-4- fluoroaniline in the presence of titanium tetrachloride and the obtained product is cyclized by the action of bistrimethylsilylacetamide and catalytic TBAF to ester-azetidinone. With alkaline hydrolysis of the ester an acid is obtained that is transformed, with the use of oxalylchloride, to an acyl chloride whose reaction with a Grignard reagent in the presence of ZnCl2 and an acetate of a transitional metal, such as e.g. palladium, produces a ketone. The latter is asymmetrically reduced with a diborane in the presence of a chiral ligand and finally the protective (9-benzyl group is hydrogenolyzed on a palladium catalyst. Again, in this case a considerable disadvantage consists in the repeated use of expensive catalysts of the palladium type as well as the repeated use of the toxic oxalylchloride.
Another common disadvantage of the above mentioned methods is the fact that complete removal of palladium is hardly feasible in practice.
Disclosure of Invention
This invention describes a new method of manufacturing (3R,4S)-l-(4-fluorophenyl)- 3 - [(3 S)-3 -(4-fluorophenyl)-3-hydroxypropyl)] -4-(4-hydroxyphenyl)-2-azetidinone (ezetirηibe) of formula I

starting from the optically active (^-iV-acyl-oxazolidide of formula II

which is reacted with an alkyleneglycol of general formula III
HO ^(CH2)nOH III
wherein n = 1 or 2, in the presence of an acidic catalyst in a hot, water-immiscible solvent (stage 1), and the obtained acetal-oxazolidide of general formula IV

wherein n means the same as above, is subjected to a reaction with a silyl-imine of general formula V

wherein R means a trialkylsilyl protective group, in the presence of a Lewis acid, e.g. titanium tetrachloride or isopropoxy titanium chloride, and a strong organic base in an inert organic solvent in the temperature range of -40 to 0 °C (stage 2), and the produced amino-oxazolidide of general formula VI

wherein both R and n have the above mentioned meanings, is cyclized by the action of a silylation agent and a catalytic quantity of a fluoride in the environment of an inert organic solvent in the temperature range of -20 to 50 0C (stage 3), and the obtained silylated azetidinone of general formula VII

wherein both R and n mean the same as above, is desilylated by the action of a desilylation agent such as tetrabutylammonium fluoride in an inert organic solvent in the temperature range of -10 to +40 0C (stage 4), and thus obtained ketal of general formula VIII
 wherein n means the same as above, is deketalized by the action of acidic agents in a mixture of water and a water-miscible solvent in the range of temperatures of 0 to 80°C (stage 5), and the obtained ketone of formula IX
wherein n means the same as above, is deketalized by the action of acidic agents in a mixture of water and a water-miscible solvent in the range of temperatures of 0 to 80°C (stage 5), and the obtained ketone of formula IX

is finally reduced with asymmetric boron agents in an inert organic solvent in the range of temperatures of -20 to +50 0C (stage 6).
We have found out that ezetimibe can be produced with the use of a method that does not require the use of expensive catalysts of the palladium type, thus eliminating the risk of retention of small quantities of this metal in the produced substance. The production procedure consists of six stages, which are further described in a detailed way.
Stage 1. (ιS)-7V-acyl-oxazolidide of formula II is reacted with an alkyleneglycol of general formula III wherein n = 1 or 2 in the presence of an acidic catalyst in a hot, water-immiscible solvent. The reaction is preferably performed with ethylenglycol (III, n = 1) in the presence of an acidic catalyst such as an organic sulfonic acid, e.g. p-toluenesulfonic acid, the inert solvents used being, e.g., benzene, toluene, xylene, 1,2-dichloroethane, preferably toluene, and beneficially at the boiling temperature of the mixture.
The compound of general formula IV can also be prepared in such a way that oxazolidide of formula II is reacted with the bis(trimefhylsilyl) derivative of the alkyleneglycol in the
presence of a catalyst, e.g. trimethylsilyl triflate. The above mentioned bis(trimethylsilyl) derivative of alkyleneglycol can be obtained by sylilation of the alkyleneglycol of formula III with trimethylsilylchloride in the presence of a suitable base, e.g. triethylamine.
Stage 2. The acetal-oxazolidide of general formula IV, wherein preferably n = 1, is subjected to a reaction with the silyl-imine of the general formula V wherein R means a trialkylsilyl protective group, such as the tert-butyldimethylsilyl, triethylsilyl or trimethylsilyl groups, preferably the tert-butyldimethylsilyl group, in the presence of a Lewis acid, e.g. titanium tetrachloride or titanium isopropoxide chloride, in the quantity of 1 to 2 equivalents, preferably 1.1 to 1.5 equivalents. The addition is performed in the presence of a strong organic base, preferably diisopropylethylamine, in the quantity of 2 to 5 equivalents, in an inert organic solvent such as dichloromethane, dichloroethane, toluene, tert-butylmethylether, tetrahydrofuran, in the temperature range of -40 to 0 0C, preferably at -35 to -20 0C. The procedure can be beneficially carried out with the use of the one-pot method in such a way that the silyl-imine of formula V is first prepared by silylation of the imine V (R = H) using a procedure known per se, and then the acetal-oxazolidide of formula IV is added onto it in the above mentioned way.
Stage 3. The amino-oxazolidide of general formula VI, wherein both R and n have the above mentioned meaning, preferably n = 1, R = TBS, is cyclized by the action of a silylation agent, such as bis-(trimethylsilyl)acetamide, and a catalytic quantity of a fluoride, preferably tetrabutylammonium fluoride. The cyclization is carried out in an inert organic solvent, such as tert-butylmethylether, tetrahydrofuran, toluene or dichloromethane, in the temperature range of -20 to 50 0C, preferably at -5 to +10 0C.
Stage 4. The silylated azetidinone of general formula VII, wherein both R and n mean the same as above, is desilylated by the action of a desilylation agent, such as tetrabutylammonium fluoride, in an inert organic solvent, as e.g. tetrahydrofuran or tert- butylmethylether, in the temperature range of -10 to +40 0C, preferably at -5 to +15 0C.
In a beneficial alternative of the procedure the product of stage 3 is not isolated but desilylation is carried out immediately, as mentioned in stage 4 (one-pot transformation).
Stage 5. The ketal of general formula VIII wherein n means the same as above is hydro lyzed by the action of acidic agents such as p-toluenesulfonic acid, methanesulfonic acid or acetic acid, in a mixture of water and a water-miscible solvent such as tetrahydrofuran, acetone, or isobutymethylketone, in the temperature range of 0 to 80 °C, beneficially at 50 to 70 °C.
Stage 6. The ketone of formula IX is reduced asymmetrically with the use of boron agents, such as e.g. a diborane, in the presence of the chiral ligand (i?)-2-methyl-CBS-oxazaborolidine in the quantity of 1 to 20 mol% in an inert organic solvent, e.g. tetrahydrofuran, tert- butylmethylether, toluene, or dichloromethane, in the temperature range of -20 to +40 °C. The obtained compound of formula I (ezetimibe) is finally purified by crystallization from a mixture of water and an alcohol, e.g. 2-propanol or methanol.
The following examples illustrate the generality of the production method of the invention but not limit the same in any way.
Example 1
Preparation of compound of formula IV (n = 1)
To a solution of (,S)-oxazolidide of formula II (15.0 g, 42.2 mmol) in 200 ml toluene there was added ethyleneglycol (formula III, n = 1) (13.1 g, 0.21 mol) andj9-toluenesulfonic acid (0.08 g, 0.42 mmol; 0.01 equiv), and the mixture was refluxed under azeotropic removal of water until separation of water had ceased (20-24 h). After cooling to 50 °C, the mixture was washed with saturated solution of sodium carbonate. Toluene layer was dried over sodium sulfate (15 min) and, after filtration of drying agent, was concentrated on rotary evaporator to 1/4 original weight. The concentrate was heated to 7O0C and then hexane was added until cloudy (60 ml). The resulting mixture might be seeded and was left to crystallize first 30 min at laboratory temperature, then 2 h at 0 °C. The separated crystals were filtered off, washed with 20 ml hexane and dried. 16.0 g, i.e. 94,9 % of acetal-oxazolidide of formula IV (n = 1) was obtained as white crystals that could be recrystallized from EtOH when necessary.
Crystallization from EtOH
Acetal-oxazolidide of formula IV (n = 1) (15.7 g) was heated with 100 ml ethanol at 80°C until dissolution (about 5-15 min). The resulting solution was left cooling to laboratory temperature under stirring, and was kept at that temperature for 3 h. Acetal-oxazolidide of
formula IV (n = 1) was filtered, washed with 10 ml ethanol and dried. Yield was in the range 82-85 %. Mp. 99-101 0C.
Preparation of compound of formula VI (n = 1, R = TBS) To a stirred mixture of imine of formula V (R = H) (2.15 g, 10.0 mniol) and tert- butyldimethylsilyl chloride (1.63 g, 10.5 mmol; 1.05 equiv) in dichloromethane (20 ml) there was added during 5 min diisopropylethylamine (5.95 ml, 34.4 mmol) under cooling to 0 0C. The mixture was left to warm up slowly to 10 0C. After 40 min, ketal of formula IV (n = 1) (3.20 g, 8.0 mmol) was added. The mixture was cooled to -33°C, and at that temperature was added 1 M solution of TiCl4 in CH2Cl2 (10 ml, 10.0 mmol) during 30 min. The mixture was stirred at -30 0C 2.5 h, and then 10% hydrochloric acid (30 ml) was added. The reaction mixture was intensely stirred 1 h at 5 °C. The separated product was filtered off, and the filtrate was combined with toluene (30 ml) and stirred at room temperature for 1 h. Separated product was filtered off. Solid portions were combined, stirred with EtOH and, after 10 min filtered, washed with EtOH and dried.
Yield: 39.8 % of compound of formula VI (n = 1, R = TBS).
Preparation of compound of formula VIII (n = 1)
To a stirred amino-oxazolidide of formula VI (n = 1, R = TMS) (2.54 g, 3.48 mmol) in THF (25 ml) there was added BSA (1.6 ml, 6.4 mmol; 1.8 equiv) and then TBAF (0.07 g, 0.22 mmol; 0.06 equiv). The stirred mixture was warmed during 1 h from 0 to 10 °C, then more TBAF (0.67 g, 2.1 mmol; 0.61 equiv) was added and the mixture was warmed during 2 h to 19 °C (progress of the reaction was monitored by TLC). Acetic acid (2.0 ml, 35 mmol) and then water (30 ml) was added, the mixture was stirred at room temperature for 15 min, and left standing overnight at 10 °C. Crystals were filtered off, washed with MeOH (2 ml) and dried (75.1%). Processing of mother liquors provided 2nd crop (0.19 g). Yield: 1.37 g, i.e. 87% ketal VIII (n = 1).
Preparation of compound of formula IX A stirred mixture of ketal of formula VIII (n = 1) (1.14 g, 2.5 mmol) and/?-toluenesulfonic acid in a mixture of acetone (25 ml) and water (2.5 ml) was brought to 65 0C and kept at that temperature for 2 h. The reaction mixture was concentrated on rotary evaporator, and the residue was dissolved in dichloromethane (50 ml) and washed with saturated solution
NaHCO3 (30 ml). The separated organic layer was washed with brine (Ix) and evaporated to dryness on rotary evaporator. Yield: 0.93 g (90,4 %).
Preparation of compound of formula I (ezetimibe)
To a solution of compound of formula IX (0.9 g, 2.2 mmol) in dichloromethane (6.6 ml) there was added 1 M solution of (i?)-2-methyl-CBS-oxazaborolidine in toluene (0.16 ml, 0.16 mmol; 0.07 equiv). The solution was cooled to -2 °C, and then 2 M solution of BH3Me2S in THF (1.6 ml, 3.2 mmol; 1.45 equiv) was added during 10 min. The mixture was stirred for 2 h at 0 °C, while the progress of the reaction was monitored by TLC. More 2 M solution of BH3Me2S in THF (0.5 ml, 1.0 mmol; 0.45 equiv) was added. After 1 h stirring at 5 0C methanol was added (0.6 ml) and the mixture was evaporated on rotary evaporator. The residue was extracted into tert-butylmethyl ether (2x 10 ml), organic layer was washed with water (Ix) and evaporated on rotary evaporator. The residue was crystallized from aqueous i- PrOH (1:1, 5 ml).
Yield: 0.51 g, i.e. 56.8 % compound of formula I.
Example 2
Preparation of compound of formula IV (n = 1) a) To a stirred suspension of compound II (n = 1) (68.9 g; 0.194 mol) in toluene (350 ml) there was added l,2-5w(trimethylsilyloxy)ethane (57.0 ml, 0.232 mol, 1.2 equiv) and trimethylsilyl triflate (3.51 ml; 19.4 mmol, 10 mol%) at room temperature. Then the mixture was brought to 50°C and left reacting for 3 to 4 h. The reaction mixture was decomposed by addition of saturated aqueous solution NaHCO3 (250 ml), after stirring while warm the organic layer was separated and aqueous layer was extracted with toluene (50 ml). Combined organic was dried (Na2SO4) and evaporated on rotary evaporator. The residue was recrystallized from ethanol, or from a mixture toluene - hexane. Mp. 98-100.5 0C. Yield: 67.8 g, i.e. 89 % compound of formula IV (n = 1).
b) To a solution of compound of formula III (n = 1) (20 g, 0.322 mol) and triethyl amine (94.4 ml, 0.677 mol) in THF (500 ml) there was added a solution of trimethylsilyl chloride (86.4 ml, 0.676 mmol) in THF (200 ml) at 0 0C during 15 min. The mixture was stirred at the same temperature for 20 min, then left to warm to room temperature and stirred for 1 h. After
filtration of crystalline compound the filtrate was evaporated on rotary evaporator. The oily residue was diluted with toluene (200 ml), compound of formula II (57.3 g, 0.161 mol) was added followed by trimethylsilyl triflate (1.46 ml, 8.05 mmol, 5 mol%). The mixture was stirred for 3 to 4 h at 50°, and then saturated aqueous solution NaHCO3 (250 ml) was added. The mixture was extracted while warm and organic layer was separated and processed further as above. Yield: 57.3 g (89 %) of compound of formula IV.
Example 3 Preparation of compound of formula V (R = TBS)
A mixture of imine of formula V (R = H) (10,0 g), tert-butyldimethylsilyl chloride (7,5 g) and imidazole (4,75 g) in DMF (45 ml) was stirred for 15 min at room temperature and left standing overnight. The reaction mixture was partitioned between toluene (120 ml) and water (120 ml), and the separated aqueous layer was extracted with toluene (40 ml). The combined organic phase was washed with water (30 ml) and evaporated. Product was crystallized from hexane. 10.1 g of silyl imine of formula V (R = TBS) was obtained. Mp. 51-53 0C.
Example 4
Preparation of compound of formula VI (n = 1, R = TBS) To a mixture of imine of formula V (R = H) (2.15 g, 10.0 mmol) and tert-butyldimethylsilyl chloride (1.58 g, 10.5 mmol; 1.05 equiv) in dichloromethane (15 ml) there was added during 5 min diisopropylethylamine (5.92 ml, 34 mmol) at 0 0C. The mixture was slowly brought to room temperature, during which time the imine had dissolved. After a total of 30 to 60 min, ketal of formula IV (n = 1) (3.20 g, 8.0 mmol) was added, the mixture was cooled to -30°C, and at that temperature was added 1 M solution of TiCl4 in CH2Cl2 (8.5 ml, 8.5 mmol) during 20 min. The mixture (dark wine-red solution) was stirred at that temperature for 2 h, and then 10% hydrochloric acid (50 ml) and toluene (70 ml) was added. The reaction mixture was intensely for another 20 min after it reached the room temperature. Separated product was filtered off, washed with EtOH (3x) and dried. Yield: 2.0 g (34 %) of compound of formula VI (n = 1 , R = TBS).
Preparation of compound of formula VI (n = 1 , R = TMS)
To a stirred mixture of imine of formula V (R = H) (2.15 g, 10.0 mmol) and trimethylsilyl chloride (1.34 ml, 10.5 mmol; 1.05 equiv) in dichloromethane (15 ml) there was added during 5 min diisopropylethyl amine (5.92 ml, 34 mmol) at 0 0C. The mixture was allowed slowly to warm to room temperature, during which imine had dissolved. After a total of 30 to 60 min, ketal of formula IV (n = 1) 3.20 g, 8.0 mmol) was added, the mixture was cooled down to -300C and, at that temperature, was added 1 M solution OfTiCl4 in CH2Cl2 (8.5 ml, 8.5 mmol) during 30 min. The mixture was stirred at the same temperature for 2.25 h, and then quenched by addition of 10% hydrochloric acid (50 ml) and toluene (70 ml) under intense stirring. The reaction mixture was intensely stirred during slow warm up to room temperature, and then for another 10 min. The separated yellow solid was filtered off, organic layer was separated, and aqueous layer was extracted with toluene (30 ml). The combined organic phase was washed with saturated solution OfNaHCO3 (Ix), dried (Na2SO4) and evaporated to dryness on rotary evaporator. Product was dissolved in a small amount of THF at reflux and then precipitated by addition of diethyl ether. The separated compound of formula VI (n = 1 ) was filtered off after cooling, washed with diethyl ether and dried. Yield: 880 mg, i.e. 32 %.
Example 5 Preparation of compound of formula VII (n = 1 , R = TMS)
To a stirred suspension of amino-oxazolidide of formula VI (n = 1, R = TMS) (1.374 g, 2.0 mmol) in tert-butylmethyl ether (20 ml) there was added BSA (1.22 ml, 5.0 mmol). The mixture was stirred for 10 min at room temperature, then cooled to 0 °C, and 0,5 M solution of TBAF in THF (0.2 ml, 0.1 mmol; 0.05 equiv) was added. The resulting mixture was stirred for 3 h at 0 0C, during which solids dissolved and progress of the reaction was monitored by TLC). After completion of the reaction acetic acid (1.0 ml, 17.5 mmol) was added, the reaction mixture was stirred at room temperature for 15 min, and then washed with saturated solution OfNaHCO3 (Ix) and water (2x). Organic phase was filtered and evaporated to dryness on rotary evaporator. Yield: 0.88 g.
Example 6
Preparation of compound of formula VIII (n = 1)
To a solution of the residue from Example 5 above (VII: n = 1, R = TMS) in THF (10 ml), there was added TBAF (0.55 g, 2.1 mmol). The mixture was stirred for 40 min at room temperature and then evaporated to dryness. Addition of MeOH to the residue caused a separation of solids. Suspension was cooled for 1 h to 10 0C, the ketal of formula VIII (n = 1) was filtered off, washed with MeOH and dried. Yield: 0.64 g, i.e. 71 %.
Example 7
Preparation of compound of formula IX
A suspension of ketal of formula VIII (n = 1) (2.63 g, 5.83 mmol) in a mixture of acetic acid (60 ml), THF (15 ml) and water (15 ml) was brought under stirring to 60 °C and kept at that temperature for 5 h, during which a homogeneous solution was formed. After completion of the reaction the mixture was concentrated on rotary evaporator and the concentrate was partitioned between toluene (100 ml) and saturated solution OfNaHCO3 (30 ml). The separated organic layer was washed with saturated solution OfNaHCO3 (Ix) and water (Ix) and, after drying (Na2SO4), evaporated to dryness on rotary evaporator. The resulting yellowish oil was dissolved in diethyl ether, and the solution again evaporated. The resulting ketal of formula IX, a white foam, could be converted by addition of hexane to a filtrable solid. Yield: 2.19 g, 92 %.
Example 8 Preparation of compound of formula I (ezetimibe)
To a solution of compound of formula IX (1.0 g, 2.455 mmol) and (i?)-2-methyl-CBS- oxazaborolidine (0.245 g, 0.245 mmol; 10 mol%) in CH2Cl2 (4 ml) there was added dropwise 2 M solution of BH3Me2S in THF (2.5 ml, 5 mmol; 2.04 equiv) during 2 h at 0 °C. Then CH2Cl2 was added (3 ml) and the mixture was stirred until disappearance of starting material (I h). The reaction was quenched by addition of MeOH (4 ml), followed by 1 N aqueous HCl (7 ml). After stirring for 10 min at 0 0C and 20 min at room temperature, the reaction mixture was concentrated on rotary evaporator. The residue was partitioned between EtOAc and water, and the separated organic layer was washed with water (Ix) and evaporated on rotary
evaporator. The residue was stirred with hexane (2 h) to give a gummy product, that could be after separation of hexane recrystallized from aqueous MeOH, or aqueous z-PrOH. Yield: 0.59 g of compound of formula I.
Claims
1. A method of manufacturing (3R,4S)-l-(4-fluorophenyl)-3-[(3S)-3-(4-fiuorophenyl)-3- hydroxypropyl)]-4-(4-hydroxyphenyl)-2-azetidinone (ezetimibe) of formula I

characterized in that in stage 1 the optically active (<S)-iV-acyl-oxazolidide of formula II

is reacted with an alkyleneglycol of general formula III
HO^^(CH2)nOH III
wherein n = 1 or 2, in the presence of an acidic catalyst in a hot, water-immiscible solvent, in stage 2 the obtained acetal-oxazolidide of general formula IV

wherein n means the same as above, is subjected to reaction with a silyl-imine of general formula V


wherein both R and n have the meanings mentioned above, is cyclized by the action of a silylation agent and the catalytic quantity of a fluoride in the environment of an inert organic solvent in the temperature range of -20 to 50 0C, in stage 4 the obtained silylated azetidinone of general formula VII

wherein both R and n have the meanings mentioned above is desilylated by the action of a desilylation agent, such as tetrabutylammonium fluoride, in an inert organic solvent in the temperature range of -10 to +40 °C, in stage 5 the thus obtained ketal of the general formula VIII


is finally reduced with asymmetrical boron agents in an inert organic solvent in the temperature range of -20 to +50 0C.
2. The method according to claim 1, characterized in that ethyleneglycol is used as the alkyleneglycol of formula III in stage 1.
3. The method according to claim 1, characterized in that p-toluenesulfonic acid in boiling toluene is used as the acidic catalyst in the hot, water-immiscible solvent in stage 1.
4. The method according to claim 1, characterized in that in stage 2, titanium tetrachloride or isopopropoxy titanium chloride in the quantity of 1 to 2 equivalents is used as the Lewis acid and diisopropylethylamine in the quantity of 2 to 5 equivalents is used as the strong organic base.
5. The method according to claims 1 and 4, characterized in that dichloromethane, toluene or tert-butylmethylether in the temperature range of -40 to +0 0C, preferably at -35 to -20 °C, are used as the inert organic solvent in stage 2. 6. The method according to claims 1, 4 and 5, characterized in that the silyl-imine of formula V is first prepared in situ in stage 2.
7. The method according to claim 1, characterized in that the cyclization in stage 3 is carried out by the action of bis-trimethylsilylacetamide and the catalytic quantity of a fluoride, preferably tetrabutylammoniurn fluoride, in an inert organic solvent, such as tert-butylmethylether, tetrahydrofuran, toluene, or dichloromethane, in the temperature range of -20 to 50 °C, preferably at -5 to +10 0C.
8. The method according to claim I5 characterized in that the desilylation in stage 4 is performed in tetrahydrofuran or tert-butylmethylether, in the temperature range of -10 to +40 0C, preferably at -5 to +15 0C.
9. The method according to claims 1, 7 and 8, characterized in that stages 3 and 4 are performed in the one-pot way.
10. The method according to claim 1, characterized in that an acidic agent, such as p- toluenesulfonic acid, methanesulfonic acid or acetic acid, in a mixture of water and a water-miscible solvent, such as tetrahydrofuran, acetone, or isobutymethylketone, in the temperature range of 0 to 80 0C, beneficially at 50 to 70 0C, is used for deacetalization in stage 5.
11. The method according to claim 1, characterized in that the reduction in stage 6 is carried out asymmetrically with boron agents such as e.g. a diborane in the presence of the chiral ligand (i?)-2-methyl-CBS-oxazaborolidine in the quantity of 1 to 20 mol% in an inert organic solvent, e.g. in tetrahydrofuran, tert-butylmethylether, toluene or dichloromethane, in the temperature range of -20 to +40 0C.
12. Acetal-oxazolidides of general formula IV

wherein n has the same meaning as in claim 1. 13. Amino-oxazolidides of general formula VI

wherein both R and n have the same meanings as in claim 1.
4. Silylated azetidinones of general formula VII
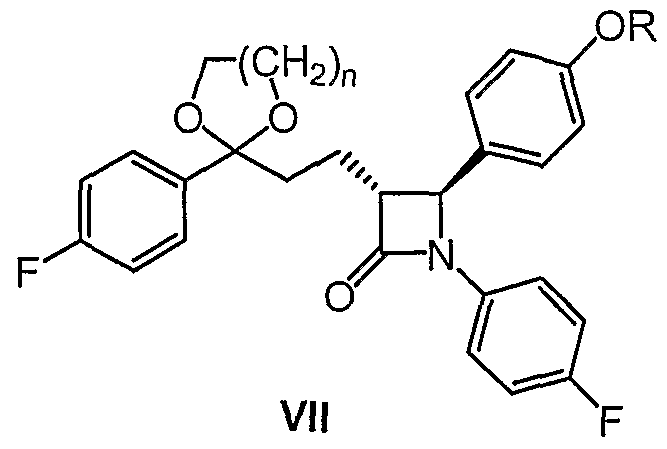
wherein both R and n have the same meanings as in claim 1.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CZ20070170A CZ302395B6 (en) | 2007-03-02 | 2007-03-02 | Process for preparing (3R,4S)-l-(4-fluorophenyl)-3-[(3S)-3-(4-fluorophenyl)-3- hydroxypropyl)]-4-(4-hydroxyphenyl)-2-azetidinone |
| CZPV-2007-170 | 2007-03-02 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2008106900A1 true WO2008106900A1 (en) | 2008-09-12 |
| WO2008106900A8 WO2008106900A8 (en) | 2008-11-06 |
Family
ID=39432207
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CZ2008/000023 WO2008106900A1 (en) | 2007-03-02 | 2008-03-03 | Method of manufacturing (3r, 4s) -1- (4-fluorophenyl) -3- [ (3s) -3- (4 -fluorophenyl) -3-hydroxypropyl) ] -4- (4-hyd roxyphenyl) -2-azetidinone |
Country Status (2)
| Country | Link |
|---|---|
| CZ (1) | CZ302395B6 (en) |
| WO (1) | WO2008106900A1 (en) |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012004382A1 (en) | 2010-07-09 | 2012-01-12 | Moehs Iberica S.L. | New method for preparing ezetimibe |
| US8178665B2 (en) * | 2005-12-20 | 2012-05-15 | Richter Gedeon Nyrt. | Process for the production of ezetimibe and intermediates used in this process |
| WO2012076030A1 (en) * | 2010-12-10 | 2012-06-14 | Pharmathen S.A. | Process for the preparation of intermediate compounds useful in the preparation of ezetimibe |
| CN102731489A (en) * | 2011-04-11 | 2012-10-17 | 天津药物研究院 | Preparation method of key ezetimibe intermediate |
| CN103159751A (en) * | 2011-12-13 | 2013-06-19 | 重庆华邦胜凯制药有限公司 | A preparation method for the derivates of phenylpyruvic acid amide ketals |
| CN104003921A (en) * | 2014-05-06 | 2014-08-27 | 南通常佑药业科技有限公司 | Preparation method of ezetimibe intermediate |
| CN104513187A (en) * | 2015-01-09 | 2015-04-15 | 安润医药科技(苏州)有限公司 | Ezetimibe synthesis method and Ezetimibe intermediate synthesis method |
| US9388440B2 (en) | 2009-04-01 | 2016-07-12 | Mylan Laboratories Limited | Enzymatic process for the preparation of (S)-5-(4-fluoro-phenyl)-5-hydroxy-1morpholin-4-yl-pentan-1-one, an intermediate of Ezetimibe and further conversion to Ezetimibe |
| CN107118144A (en) * | 2016-02-24 | 2017-09-01 | 上海医药工业研究院 | The reduction preparation technology of ezetimibe and its intermediate |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5886171A (en) * | 1996-05-31 | 1999-03-23 | Schering Corporation | 3-hydroxy gamma-lactone based enantioselective synthesis of azetidinones |
| WO2005113495A1 (en) * | 2004-05-21 | 2005-12-01 | Sanofi-Aventis Deutschland Gmbh | Method for producing diphenyl azetidinone derivatives |
| WO2006137080A1 (en) * | 2005-06-22 | 2006-12-28 | Manne Satyanarayana Reddy | Improved process for the preparation of ezetimibe |
| WO2007072088A1 (en) * | 2005-12-20 | 2007-06-28 | Richter Gedeon Nyrt. | Process for the production of ezetimibe and intermediates used in this proces |
| WO2007119106A2 (en) * | 2005-12-22 | 2007-10-25 | Medichem, S.A. | Processes for preparing intermediate compounds useful for the preparation of ezetimibe |
-
2007
- 2007-03-02 CZ CZ20070170A patent/CZ302395B6/en not_active IP Right Cessation
-
2008
- 2008-03-03 WO PCT/CZ2008/000023 patent/WO2008106900A1/en active Application Filing
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5886171A (en) * | 1996-05-31 | 1999-03-23 | Schering Corporation | 3-hydroxy gamma-lactone based enantioselective synthesis of azetidinones |
| WO2005113495A1 (en) * | 2004-05-21 | 2005-12-01 | Sanofi-Aventis Deutschland Gmbh | Method for producing diphenyl azetidinone derivatives |
| WO2006137080A1 (en) * | 2005-06-22 | 2006-12-28 | Manne Satyanarayana Reddy | Improved process for the preparation of ezetimibe |
| WO2007072088A1 (en) * | 2005-12-20 | 2007-06-28 | Richter Gedeon Nyrt. | Process for the production of ezetimibe and intermediates used in this proces |
| WO2007119106A2 (en) * | 2005-12-22 | 2007-10-25 | Medichem, S.A. | Processes for preparing intermediate compounds useful for the preparation of ezetimibe |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8178665B2 (en) * | 2005-12-20 | 2012-05-15 | Richter Gedeon Nyrt. | Process for the production of ezetimibe and intermediates used in this process |
| US9388440B2 (en) | 2009-04-01 | 2016-07-12 | Mylan Laboratories Limited | Enzymatic process for the preparation of (S)-5-(4-fluoro-phenyl)-5-hydroxy-1morpholin-4-yl-pentan-1-one, an intermediate of Ezetimibe and further conversion to Ezetimibe |
| WO2012004382A1 (en) | 2010-07-09 | 2012-01-12 | Moehs Iberica S.L. | New method for preparing ezetimibe |
| WO2012076030A1 (en) * | 2010-12-10 | 2012-06-14 | Pharmathen S.A. | Process for the preparation of intermediate compounds useful in the preparation of ezetimibe |
| CN102731489A (en) * | 2011-04-11 | 2012-10-17 | 天津药物研究院 | Preparation method of key ezetimibe intermediate |
| CN102731489B (en) * | 2011-04-11 | 2016-10-26 | 天津药物研究院有限公司 | A kind of preparation method of key ezetimibe intermediate |
| CN103159751A (en) * | 2011-12-13 | 2013-06-19 | 重庆华邦胜凯制药有限公司 | A preparation method for the derivates of phenylpyruvic acid amide ketals |
| CN104003921A (en) * | 2014-05-06 | 2014-08-27 | 南通常佑药业科技有限公司 | Preparation method of ezetimibe intermediate |
| CN104513187A (en) * | 2015-01-09 | 2015-04-15 | 安润医药科技(苏州)有限公司 | Ezetimibe synthesis method and Ezetimibe intermediate synthesis method |
| CN107118144A (en) * | 2016-02-24 | 2017-09-01 | 上海医药工业研究院 | The reduction preparation technology of ezetimibe and its intermediate |
Also Published As
| Publication number | Publication date |
|---|---|
| CZ2007170A3 (en) | 2008-10-22 |
| WO2008106900A8 (en) | 2008-11-06 |
| CZ302395B6 (en) | 2011-04-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2008106900A1 (en) | Method of manufacturing (3r, 4s) -1- (4-fluorophenyl) -3- [ (3s) -3- (4 -fluorophenyl) -3-hydroxypropyl) ] -4- (4-hyd roxyphenyl) -2-azetidinone | |
| JP3640888B2 (en) | Synthesis process of azetidinone | |
| EP1971573B1 (en) | Processes for preparing intermediate compounds useful for the preparation of ezetimibe | |
| EP1362855B1 (en) | Beta-lactam compounds, process for repoducing the same and serum cholesterol-lowering agents containing the same | |
| JP2008540557A (en) | Process for producing phenol-type 4-biphenylylazetidin-2-one | |
| CA2630737A1 (en) | Process for the production of ezetimibe and intermediates used in this process | |
| KR101156588B1 (en) | Method of preparing ezetimibe and intermediates used therein | |
| JP3233403B2 (en) | Optically active intermediate and method for producing the same | |
| JP2005516064A (en) | Method for producing organic compound | |
| WO2009067960A2 (en) | A method of manufacturing (3r,4s)-l-(4-fluorophenyl)-3-[(3s)-3-(4-fluorophenyl)-3- hydroxypropyl)]-4-(4-hydroxyphenyl)-2-azetidinone and its intermediates | |
| WO2009140932A2 (en) | Method of producing (3r,4s)-l-(4-fluorophenyl)-3-[(3s)-3-(4-fluorophenyl)- hydroxypropyl)]-4-(4-hydroxyphenyl)-2-azetidinone | |
| JP3252032B2 (en) | Method for producing β-methylcarbapenem intermediate | |
| JPWO2008020597A1 (en) | Method for producing 1-methylcarbapenem intermediate | |
| JP2010524923A (en) | Stereoselective production method of 4-BMA using chiral auxiliary | |
| JP4135639B2 (en) | NOVEL ORGANIC SILICON COMPOUND, OPTICALLY ACTIVE FORM, METHOD FOR PRODUCING THE ORGANIC SILICON COMPOUND, AND USE THEREOF | |
| JP3748933B2 (en) | Process for producing 1-substituted azetidinone derivatives | |
| JP2608458B2 (en) | Method for producing 4-acetoxyazetidinone derivative | |
| JP5059355B2 (en) | Method for producing oxazolidine derivative | |
| KR100502833B1 (en) | Improved preparation method of simvastatin and their intermediates | |
| JPH0657683B2 (en) | Optically active amino acid derivative | |
| JP4747748B2 (en) | Preparation of imidazopyran derivatives | |
| KR100283608B1 (en) | Method of preparing 1-betamethyl-2-formyl carbapenem derivatives | |
| JP5143556B2 (en) | Novel synthesis of carbapenem synthesis intermediates using sugar templates | |
| JP2893473B2 (en) | Process for producing (+)-equilenin and intermediate | |
| JPH0321025B2 (en) |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08715424 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 08715424 Country of ref document: EP Kind code of ref document: A1 |